Okay, so I'm working on implementing the semi-empirical method PM6 (by Jimmy "Mopac" Stewart) in GAMESS-US.
The status is; (before I started working) GAMESS has up to and including PM3 already implemented. So the idea is just to update the SE parameters and substitute the subroutines necessary to get PM6 working. Without prior knowledge to GAMESS this really did not sound like a big deal, as the differences between PM6 and PM3 only lies in the way the parameters are used (roughly). The parameterization of PM3 (and AM1) is utilised in the core-core repulsion term (nuclear repulsion) of the Heat of Formation to compensate for the aproximations made in SE methods. Heat of Formation is calcuated acordingly:
The emperical parameters from PM3 is fitted via a scaleing factor on the core-core term to fit the experimental heat of formation for the molecule. Fitting of the data and derivation of the parameters was done my Jimmy Stewart, for his program MOPAC where the methods were original implemented. The PM3 core-core repulsion term looks like this;
which is then summed over all nuclear repulsions/interactions between any atom A and B. This core-core term needs to be substituted with the new term from PM6:
Note that the $\alpha$ parameter is now a di-atomic parameter unlike the mono-atomic parameter is PM3. Another parameter $x$ is also introduced, but that is 'pritty much it'. (there are also a Lennard Jones Term and a van der Waals term, but that is for another blog post). The parameters are all located in the PM6 article, but Jimmy Stewart was kind of enough to send his files including his implementation of PM6 core equation and the list of all parameters. This saved me alot of pointless typing time, so thanks!
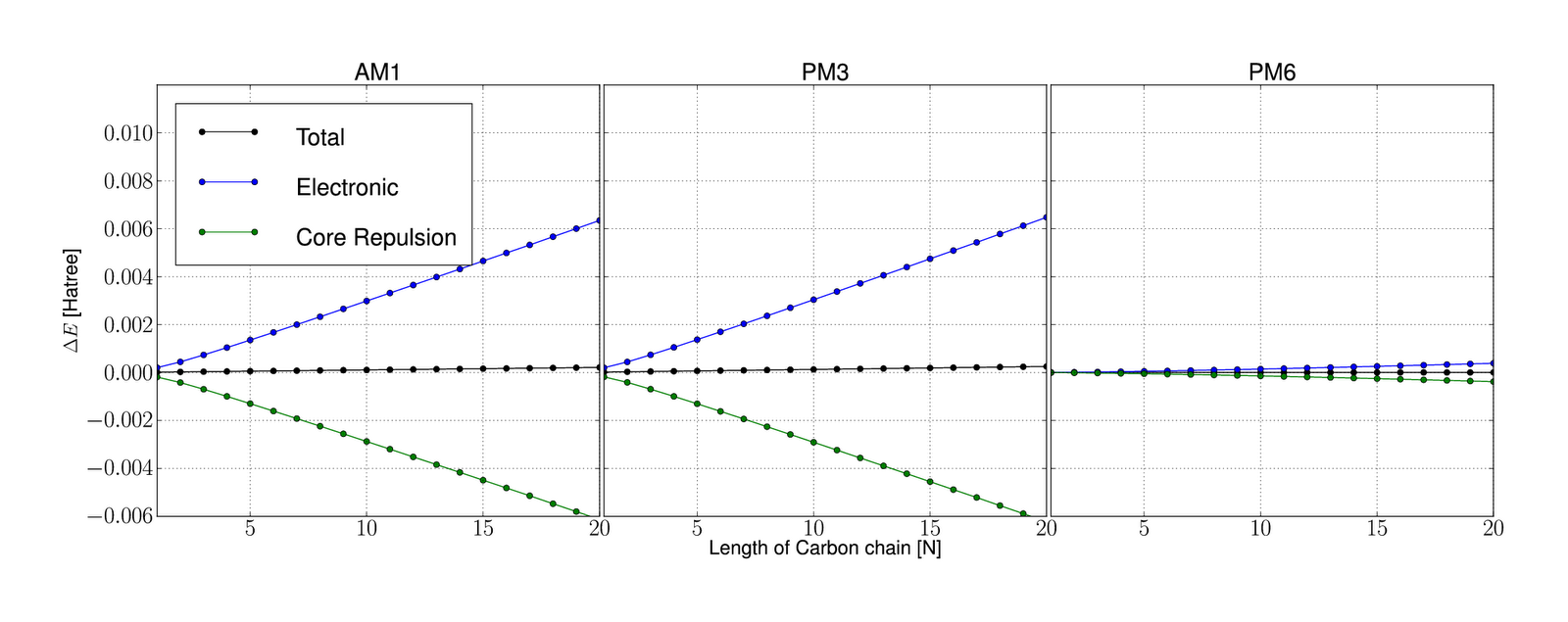
Okay, so after implementing the new PM6 specific code and the corresponding parameters, I discovered that the result did not match its MOPAC equivalent. In fact nether electronic, core or the total energy fit the MOPAC value. This was happening for single point energy calculation even for very small molecules (even water). For reference I did a similar test for PM3 and AM1, and found that the already implemented methods results did not fit it's rightfull energies (MOPAC energy) with the same order of magnitude as PM6, which was quickly discovered to be size dependt. This is clearly shown in the below figure, which shows single point energy calculations on a carbon chain from 1 carbon to 20 carbons
.

Energy difference $\Delta E$ is calculated from mopac energy minus the corresponding gamess energy.
Arrgghh! How am I going to implement a new method, when the already implemented methods varies this much from the original program?
Okay, so the problem was that the SE part of GAMESS was based on a very old version of MOPAC, and so we figured alot of the energy deviation must be originating from the lack of update on physical constants. The MOPAC integrals use two physical constants to calculate the integrals in atomic units, namely bohr radius and electron volts, so by using grep I found all the places where the constants/variables were defined (which was alot!), and then updated acording to the constant defined on MOPAC's website using a common block, instead of alot of local instances.
This resulted in

Okay, but is this better? Hell yeah! The total energy is clearly more stable compared to MOPAC energy, which is the energy that matters most. The deviation in the nuclear and electronic energy looks very much linear which hints to more constants needed to be updated. Note I have only updated the constants located in MOPAC part of GAMESS and therefor only effects the semi-empirical part of GAMESS.
However the effect is there, and even though the energy is working now, it will prove a problem for people who wants to reproduce data already calculated with GAMESS. So be warned GAMESS users, keep a copy of your GAMESS when the PM6 update is integrated in GAMESS-US.
PM6 Gradient:
The integration of gradient was actually really easy, because GAMESS only uses numerical gradients for semi-emperical calculations.
Am I done? Unfortunately no. To get PM6 fully working I need to implement the d-integrals from MOPAC. As it is now only s- and p-orbitals are used for calculating the integrals. Is that easy? No.
To be continued...
tldr; PM6, PM3 and AM1 did not work as expected, which was partially fixed by updating physical constants in the semi-empirical part of GAMESS. PM6 energy and gradient now works up to including Ne, but will need d-orbitals before it is fully operational.
The status is; (before I started working) GAMESS has up to and including PM3 already implemented. So the idea is just to update the SE parameters and substitute the subroutines necessary to get PM6 working. Without prior knowledge to GAMESS this really did not sound like a big deal, as the differences between PM6 and PM3 only lies in the way the parameters are used (roughly). The parameterization of PM3 (and AM1) is utilised in the core-core repulsion term (nuclear repulsion) of the Heat of Formation to compensate for the aproximations made in SE methods. Heat of Formation is calcuated acordingly:
$$\Delta H_f = E_{\rm Elect} + E_{\rm Core} - \sum_{A}^{} E_{el}^{A} + \sum_{A}^{} \Delta H_{f}^{A}$$
The emperical parameters from PM3 is fitted via a scaleing factor on the core-core term to fit the experimental heat of formation for the molecule. Fitting of the data and derivation of the parameters was done my Jimmy Stewart, for his program MOPAC where the methods were original implemented. The PM3 core-core repulsion term looks like this;
$$E_n(A,B) = Z_A Z_B \langle s_A s_A | s_B s_B \rangle \left ( 1 + e^{-\alpha_A R_{AB}} + e^{-\alpha_B R_{AB}} \right ) $$
which is then summed over all nuclear repulsions/interactions between any atom A and B. This core-core term needs to be substituted with the new term from PM6:
$$E_n(A,B) = Z_A Z_B \langle s_A s_A | s_B s_B \rangle \left ( 1 + x_{AB} e^{-\alpha_{AB} (R_{AB} + 3 \cdot 10^{-4} R_{AB}^6)} \right )$$
Note that the $\alpha$ parameter is now a di-atomic parameter unlike the mono-atomic parameter is PM3. Another parameter $x$ is also introduced, but that is 'pritty much it'. (there are also a Lennard Jones Term and a van der Waals term, but that is for another blog post). The parameters are all located in the PM6 article, but Jimmy Stewart was kind of enough to send his files including his implementation of PM6 core equation and the list of all parameters. This saved me alot of pointless typing time, so thanks!
Okay, so after implementing the new PM6 specific code and the corresponding parameters, I discovered that the result did not match its MOPAC equivalent. In fact nether electronic, core or the total energy fit the MOPAC value. This was happening for single point energy calculation even for very small molecules (even water). For reference I did a similar test for PM3 and AM1, and found that the already implemented methods results did not fit it's rightfull energies (MOPAC energy) with the same order of magnitude as PM6, which was quickly discovered to be size dependt. This is clearly shown in the below figure, which shows single point energy calculations on a carbon chain from 1 carbon to 20 carbons
.

Energy difference $\Delta E$ is calculated from mopac energy minus the corresponding gamess energy.
Arrgghh! How am I going to implement a new method, when the already implemented methods varies this much from the original program?
Okay, so the problem was that the SE part of GAMESS was based on a very old version of MOPAC, and so we figured alot of the energy deviation must be originating from the lack of update on physical constants. The MOPAC integrals use two physical constants to calculate the integrals in atomic units, namely bohr radius and electron volts, so by using grep I found all the places where the constants/variables were defined (which was alot!), and then updated acording to the constant defined on MOPAC's website using a common block, instead of alot of local instances.
This resulted in
Okay, but is this better? Hell yeah! The total energy is clearly more stable compared to MOPAC energy, which is the energy that matters most. The deviation in the nuclear and electronic energy looks very much linear which hints to more constants needed to be updated. Note I have only updated the constants located in MOPAC part of GAMESS and therefor only effects the semi-empirical part of GAMESS.
However the effect is there, and even though the energy is working now, it will prove a problem for people who wants to reproduce data already calculated with GAMESS. So be warned GAMESS users, keep a copy of your GAMESS when the PM6 update is integrated in GAMESS-US.
PM6 Gradient:
The integration of gradient was actually really easy, because GAMESS only uses numerical gradients for semi-emperical calculations.
Am I done? Unfortunately no. To get PM6 fully working I need to implement the d-integrals from MOPAC. As it is now only s- and p-orbitals are used for calculating the integrals. Is that easy? No.
To be continued...
tldr; PM6, PM3 and AM1 did not work as expected, which was partially fixed by updating physical constants in the semi-empirical part of GAMESS. PM6 energy and gradient now works up to including Ne, but will need d-orbitals before it is fully operational.
No comments:
Post a Comment